
This post takes a look at an extension of the standard joint longitudinal-survival model, which is to incorporate competing risks. Let’s start by formally defining the model.
We will assume a continuous longitudinal outcome,
$$y_{i}(t) = m_{i}(t) \epsilon_{i}(t)$$
where
$$m_{i}(t) = X_{1i}(t)\beta_{1} + Z_{i}(t)b_{i}$$
and \(\epsilon_{i}(t)\) is our normally distributed residual variability. We call \(m_{i}(t)\) our trajectory function, representing the true underlying value of the continuous outcome at time t, which is a function of fixed and random effects with associated design matrices, \(X_{2i}(t)\) and \(Z_{i}(t)\), and coefficients, \(\beta_{2}\) and \(b_{i}\). We assume normally distributed random effects,
$$b_{i} \sim N(0,\Sigma)$$.
We then have K competing events, each of which can be dependent on some (or many) aspects of the biomarker trajectory function, \(m_{i}(t)\). To keep things simple, we will work with the current value association structure. So, for the kth competing risk we parameterise the cause-specific hazard function as,
$$h_{ik}(t) = h_{0k}(t) \exp (X_{2ik}\beta_{2k} + \alpha_{k}m_{i}(t))$$
where \(\alpha_{k}\) is the log hazard ratio for the kth cause, for a one-unit increase in the biomarker, at time t. Each competing event can be modelled however we like, with different baseline hazard functions, and with different covariate effects. Of course, we can also have different or additional association structures for each event.
To illustrate, we’re going to show you how to simulate data from a joint longitudinal and competing risks model, with two competing risks, representing death from cancer and death from other causes, and then show how to fit the true model using merlin.
Let’s assume a simple random intercept and random linear trend for the biomarker trajectory, i.e.
$$m_{i}(t) = (\beta_{10} + b_{0i}) + (\beta_{11} + b_{1i})t$$
We start by setting a seed (as we always should), generate a sample of 300 patients, and create an id variable.
. clear
. set seed 7254
. set obs 300
Number of observations (_N) was 0, now 300.
. gen id = _nNext we generate our subject-specific random effects, for the intercept and slope. We need a single draw from each distribution, per patient. We’re assuming the random effects are independent, but that’s easily adapted using drawnorm. We also generate a treatment group indicator, assigning to each arm with 50% probability.
. gen b0 = rnormal(0,1)
. gen b1 = rnormal(0,0.5)
. gen trt = runiform()>0.5Now we simulate cause-specific event times, using survsim. Simulating joint longitudinal-survival data is not the easiest of tasks mathematically, but thanks to survsim it’s pretty simple, especially given the recent additions to its capabilities (Crowther, 2022). We simply specify our cause-specific hazard functions, and survsim does the hard work (numerical integration nested within root-finding), details in Crowther and Lambert (2013).
We specify our baseline distribution parameters, the scale and shape, for Weibull baseline hazards for causes one and two, and an administrative censoring time of 5 years. We also specify a treatment effect with a log hazard ratio of -0.5 on cause 1 and -0.2 on cause 2. Putting this all together in user-defined functions in survsim, simulating under the current value association structure, defining \(\alpha_{1}=0.2\), we get,
. //cause one
. local maxt 5
. local l1 0.1
. local g1 1.2
. local l2 0.05
. local g2 1.5
. survsim stime state event, /// new variables
> hazard1( /// cause 1 (cancer)
> user(`l1':*`g1':*{t}:^(`g1'-1) /// user-defined function
> :* exp(0.2 /// biomarker
> :* (b0 :+ (0.1:+b1):*{t}) /// trajectory
> )) ///
> covariates(trt -0.5)) /// treatment effect (PH)
> hazard2( /// cause 2 (other)
> user(`l2':*`g2':*{t}:^(`g2'-1) /// user-defined function
> :* exp( -0.2 :* /// biomarker
> (b0 :+ (0.1:+b1):*{t}) /// trajectory
> )) ///
> covariates(trt -0.2)) /// treatment effect (PH)
> maxtime(`maxt') // admin. censoring
variables stime0 to stime1 created
variables state0 to state1 created
variables event1 to event1 createdNow survsim is designed for many situations, such as allowing you to define your own custom hazard functions, which can depend on time…or a time-dependent biomarker trajectory. It also provides a general framework to simulate from competing risks and multi-state survival models. In this example, we are simulating from an all-cause hazard function, which is a function of two cause-specific hazards, each of which depends on a time-dependent biomarker trajectory. That’s a mouthful. Ok, so to start we define some new variable stubs, to contain the variables that survsim will create. stime* will contain the start and stop times for each transition, state* will contain the start and stop states, and event* will be flag indicator variables for whether an event/transition occurred, or an obervation was censored.
Let’s see what we created:
. list id trt stime* state* event* if _n<=5
+----------------------------------------------------------+
| id trt stime0 stime1 state0 state1 event1 |
|----------------------------------------------------------|
1. | 1 1 0 5 1 1 0 |
2. | 2 0 0 1.8992871 1 2 1 |
3. | 3 1 0 .9001096 1 2 1 |
4. | 4 1 0 1.0330409 1 2 1 |
5. | 5 1 0 2.9920639 1 3 1 |
+----------------------------------------------------------+So all observations started in state 1 (state0 is a 1) at time 0 (stime0 is 0). Observation 1 was censored (event1 is a 0) at 5 years, observations 2, 3 and 4 moved to state 2 at various times, i.e. died due to cancer, and observation 5 died from other causes (state1 is a 3) at 2.756 years. As it’s a competing risks situation (which is what survsim will assume unless you specify your own transition matrix), then observations can only go from the starting state 1 to states 2 or 3, corresponding to hazard1 and hazard2, respectively.
All that’s left to do is to create our cause-specific event indicators, which we will need in our estimation step. We’ll then drop variables we no longer need.
. gen byte cancer = state1==2
. gen byte other = state1==3
. drop stime0 state0 state1 event1Now we’re all done with the competing risk outcomes. Onto the biomarker.
Let’s assume a setting where our biomarker is recorded at baseline, and then annually, so each patient can have up to five measurements. The easiest thing to do is to expand our dataset, by creating replicants of each row, and then drop any observations which occur after each patient’s event/censoring time.
. expand 5
(1,200 observations created)
. bys id : gen time = _n-1
. drop if time>stime1
(465 observations deleted)We generate our oberserved longitudinal biomarker measurements from the true model above,
. gen xb = b0 + 0.1 * time + b1 * time
. gen y = rnormal(xb,0.5) //measurement errorIt’s easiest to work in wide format for merlin (I mean in terms of the outcomes, they are side by side, but within each outcome the observations are in long format), so for the competing risks outcomes we need to have only a single row of data per patient, so we replace the repeated event times and event indicators with missing values.
. bys id (time) : replace stime1 = . if _n>1
(735 real changes made, 735 to missing)
. bys id (time) : replace cancer = . if _n>1
(735 real changes made, 735 to missing)
. bys id (time) : replace other = . if _n>1
(735 real changes made, 735 to missing)We’re now all set up to fit our data-generating, true model, with merlin, using the following syntax:
merlin (y /// biomarker outcome
time /// fixed linear time
time#M2[id]@1 /// random slope on linear time
M1[id]@1, /// random intercept
family(gaussian) /// distribution
timevar(time)) /// variable representing time
(stime1 /// survival time
trt /// baseline treatment
EV[y], /// expected value of biomarker
family(weibull, /// distribution
failure(cancer)) /// cause-specific event indicator
timevar(stime1)) /// timevar
(stime1 /// survival time
trt /// baseline treatment
EV[y], /// expected value of biomarker
family(weibull, /// distribution
failure(other)) /// cause-specific event indicator
timevar(stime1)) // timevarwhich we would say is rather elegantly simple to specify for such a complex model…you may disagree. We’re using the EV[y] element type to link the current value of the biomarker to each cause-specific hazard model, making sure to specify the timevar() options, so time is correctly handled. We can use stime1 as our outcome in both survival models but simply use the cause-specific event indicators so we get the correct contributions to the likelihood. It’s important to note that competing risks models with exactly observed event data, can generallay be fitted either with a stacked dataset, or separately, with cumulative incidence predictons calculated post-estimation – this is not true in such a joint model, since the biomarker model appears in both cause-specific hazard functions, and hence must be estimated simultaneously.
Let’s fit the model:
. merlin (y time time#M2[id]@1 M1[id]@1, family(gaussian) timevar(time)) ///
> (stime1 trt EV[y], family(weibull, failure(cancer)) timevar(stime1)) ///
> (stime1 trt EV[y], family(weibull, failure(other)) timevar(stime1))
Fitting fixed effects model:
Fitting full model:
Iteration 0: log likelihood = -2438.3381 (not concave)
Iteration 1: log likelihood = -2405.6907
Iteration 2: log likelihood = -2171.3013
Iteration 3: log likelihood = -2069.3195
Iteration 4: log likelihood = -2032.6354
Iteration 5: log likelihood = -2023.3654
Iteration 6: log likelihood = -2023.1791
Iteration 7: log likelihood = -2023.1787
Iteration 8: log likelihood = -2023.1787
Mixed effects regression model Number of obs = 1,035
Log likelihood = -2023.1787
------------------------------------------------------------------------------
| Coefficient Std. err. z P>|z| [95% conf. interval]
-------------+----------------------------------------------------------------
y: |
time | .1331602 .0368502 3.61 0.000 .0609352 .2053853
time#M2[id] | 1 . . . . .
M1[id] | 1 . . . . .
_cons | .126154 .0672743 1.88 0.061 -.0057013 .2580092
sd(resid.) | .5107904 .0159339 .4804961 .5429948
-------------+----------------------------------------------------------------
stime1: |
trt | -.6689459 .181305 -3.69 0.000 -1.024297 -.3135946
EV[] | .1962076 .0564493 3.48 0.001 .085569 .3068462
_cons | -1.871467 .1573127 -11.90 0.000 -2.179794 -1.56314
log(gamma) | .0748063 .0806735 0.93 0.354 -.0833108 .2329233
-------------+----------------------------------------------------------------
stime1: |
trt | -.3152746 .2263077 -1.39 0.164 -.7588294 .1282803
EV[] | -.306471 .069009 -4.44 0.000 -.4417262 -.1712158
_cons | -3.0444 .2563032 -11.88 0.000 -3.546745 -2.542055
log(gamma) | .4395772 .097876 4.49 0.000 .2477438 .6314106
-------------+----------------------------------------------------------------
id: |
sd(M1) | 1.07923 .0509042 .9839322 1.183757
sd(M2) | .5011646 .0292952 .446914 .5620007
------------------------------------------------------------------------------The model fits beautifully…phew.
Now we have our fitted model, generally of most interest within a competing risks analysis is both the cause-specific hazard ratios, which we get from our results table, and estimates of the cause-specific cumulative incidence functions.
We use merlin‘s predict engine to help us with such things. When predicting something which depends on time, it’s often much easier to specify a timevar(), which contains timepoints at which to predict at, which subsequently makes plotting much easier. So now we’ll generate a new variable tvar which will contain time points between 0 and 5, at 500 equally spaced points.
. range tvar 0 5 500
(535 missing values generated)Let’s now predict each cause-specific cumulative incidence function, in particular, we’ll predict marginal CIFs (integrating out the random effects), so we can interpret them as population-average predictions. We’ll also use the at() option so we can investigate the effect of treatment.
. predict cif1, cif marginal outcome(2) at(trt 0) timevar(tvar)
(535 missing values generated)
. predict cif2, cif marginal outcome(2) at(trt 1) timevar(tvar)
(535 missing values generated)
. predict cif3, cif marginal outcome(3) at(trt 0) timevar(tvar)
(535 missing values generated)
. predict cif4, cif marginal outcome(3) at(trt 1) timevar(tvar)
(535 missing values generated)For plotting purposes, we often create stacked plots of CIFs, and one way to do this is to add them appropriately together to draw the area under the curve, and then overlay one of the CIFs.
. gen totalcif1 = cif1 + cif3
(535 missing values generated)
. gen totalcif2 = cif2 + cif4
(535 missing values generated)We now plot the stacked, marginal CIFs for those in the placebo group and those in the treated group, and combine (using grc1leg from SSC):
. twoway (area totalcif1 tvar)(area cif3 tvar), name(g1,replace) ///
> xtitle("Time since entry") ytitle("Cumulative incidence") ///
> title("Placebo group") legend(cols(1) ///
> order(1 "Prob. of death due to cancer" ///
> 2 "Prob. of death due to other causes")) ///
> ylabel(,angle(h) format(%2.1f)) ylabel(0(0.1)1) nodraw. twoway (area totalcif2 tvar)(area cif4 tvar), name(g2,replace) ///
> xtitle("Time since entry") ytitle("Cumulative incidence") ///
> title("Treated group") ///
> legend(cols(1) order(1 "Prob. of death due to cancer" ///
> 2 "Prob. of death due to other causes")) ///
> ylabel(,angle(h) format(%2.1f)) ylabel(0(0.1)1) nodraw. grc1leg g1 g2 //combine them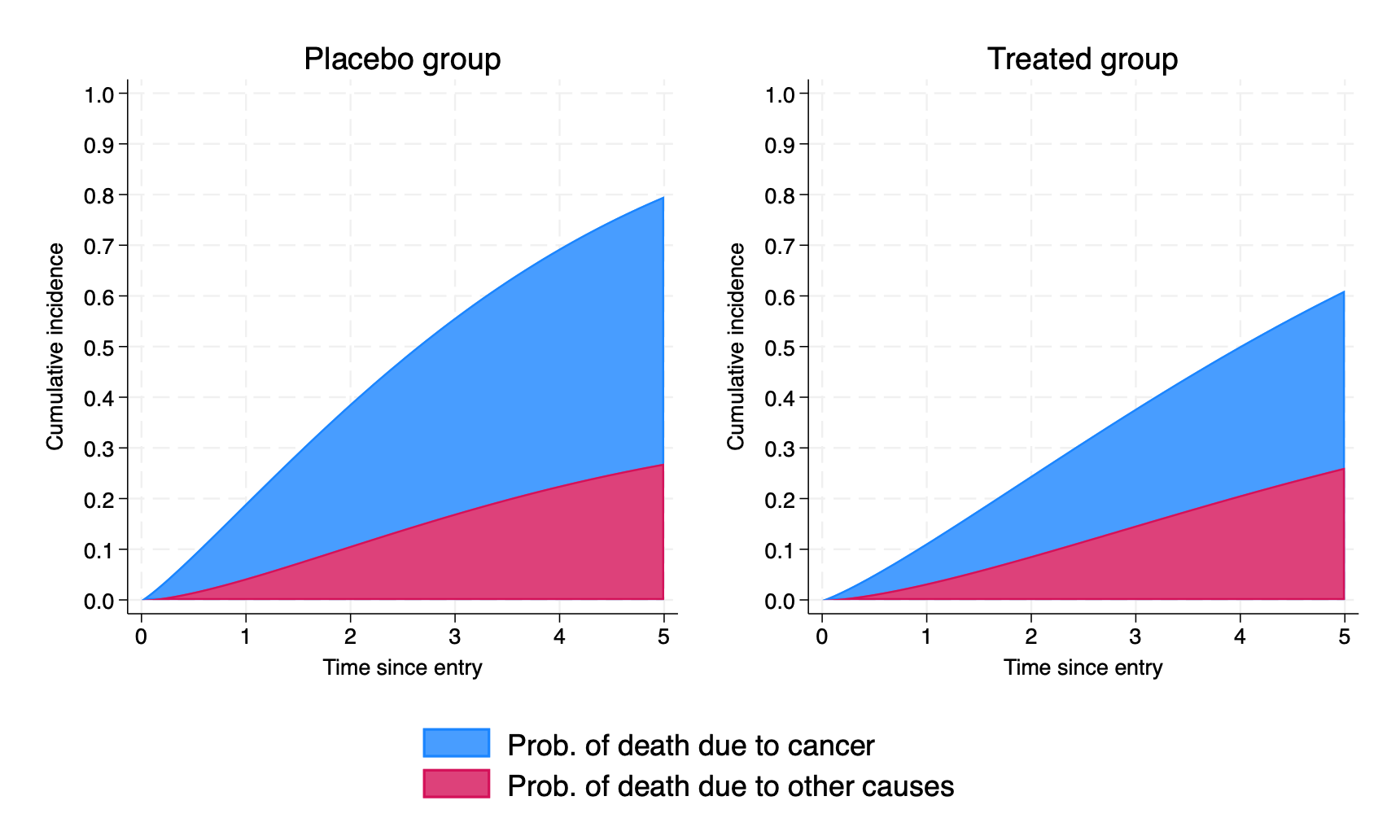
We can directly quantify the impact of treatment on each CIF by predicting the difference, using the cifdifference option, combined with at1() and at2() statements.
. predict diffcif, cifdifference marginal outcome(2) ///
> at1(trt 1) at2(trt 0) timevar(tvar) ///
> ci
(535 missing values generated)
note: confidence intervals calculated using Z critical values.. twoway (rarea diffcif_lci diffcif_uci tvar) ///
> (line diffcif tvar), name(g3,replace) ///
> xtitle("Time since entry") ///
> ytitle("Difference in cumulative incidence") ///
> title("CIF({it:t} | treated) - CIF({it:t} | placebo)") ///
> ylabel(,angle(h) format(%3.2f)) ///
> legend(order(2 "Diff. in CIF" 1 "95% confidence interval"))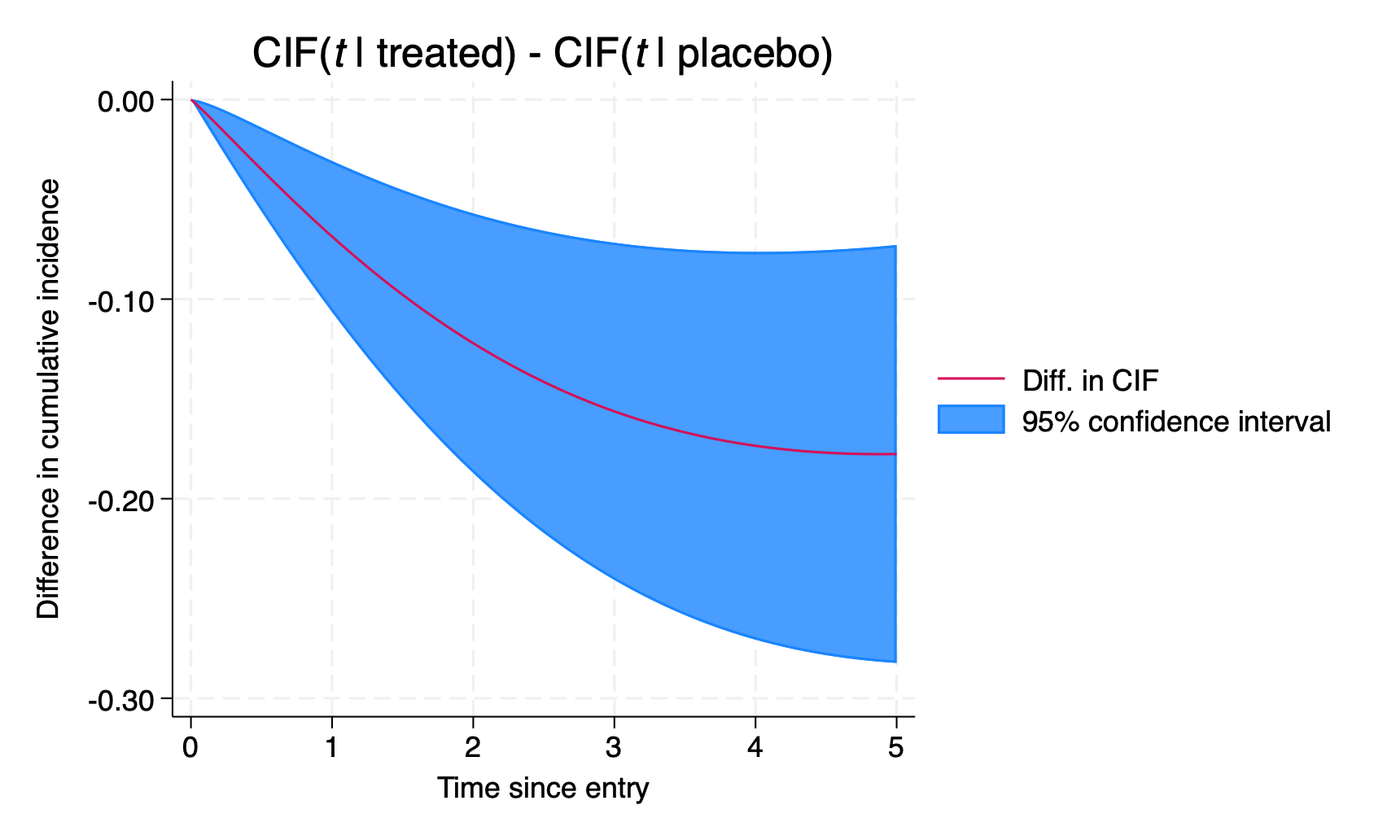
We’ll leave it there for now. The joint model we’ve covered is an introductory example in the competing risks setting. Given merlin‘s capabilities, it’s rather simple to extend to using other associations structures, such as the rate of change (dEV[y]) or the integral (iEV[y]) of the trajectory function, and to use different distributions for each cause-specific hazard model…the list of extensions goes on.
Happy joint modelling.

Specialist subjects
Clinical Trial Services

Specialist subjects
Methods Development

Specialist subjects








